
The new regulatory framework for AI / ML-enabled SaMDs is driven by two main concepts.
- Patient-centered approach incorporating usability, accountability and transparency for SaMD users,
- Total product lifecycle approach.
The regulatory framework aims to promote a “total product life cycle” (TPLC) mechanism that ultimately supports both FDA and manufacturers in providing increased benefits to patients and providers and where manufacturers are continually vigilant in maintaining the safety and effectiveness of their SaMD.
The process focuses on the software developer practices rather than on the software itself. The certification process foresees various steps:
- Pre-check approval: A robust post-market oversight
- A lighter premarket scrutiny: When needed, a new submission for significant changes.
The pre-check approval focuses on quality systems and good machine learning practices[1] and represents a kind of “excellence appraisal” of the manufacturer / developer. It is is focused on procedural safeguards like the quality of the data used, the development techniques, and the validation procedures. More specifically in the pre-check approval FDA will assess (see diagram of fig.1):
- the culture of quality and organizational excellence of a particular company,
- the quality of its software development and testing,
- and the monitoring of the performance of its products.
The rules related to change control for SaMDs are based on the FDA Quality System Regulation (21 CFR Part 820) and specifically the 21 CFR Part 820.23 (Design changes) and the 21 CFR Part 820.70 (Production and process changes).
Companies in their premarket submissions for AI products will include their plans for anticipated modifications (“SaMD Pre-Specifications” (SPS)) and the “algorithm change protocol” (ACP)) i.e. the processes the company will follow to make those changes.
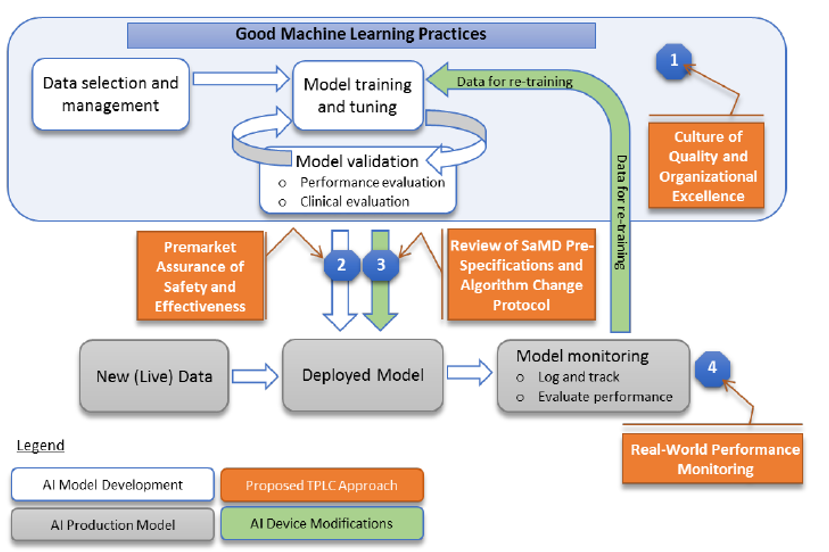
Fig.1 – Good ML practices with integrated a “total product life cycle TPLC” approach
- Conduct a lighter premarket scrutiny for those SaMDs that require premarket submission; the intent is to demonstrate reasonable assurance of safety and effectiveness and establish clear expectations for manufacturers of AI/ML-based SaMD to continually manage patient risks throughout the lifecycle.
- A robust post-market oversight once the algorithms enter into clinical care; it will provide a proof in a “clinical, real world environment”. An increased transparency between manufacturers / SW developers and FDA is fostered with the goal of maintaining continued assurance of safety and effectiveness.
- When needed, a new submission for significant changes
Certain changes, like improvements in performance and changes in data inputs, would not require a new submission, but others, like significant changes to the intended use, would. The need of a new submission is defined in the FDA guidelines “Deciding When to Submit a 510(k) for a Software Change to an Existing Device”.
The classification of SaMD
SaMDs are classified according to a risk-based approach taking into account the intended use and the healthcare situation. More specifically the intended use of the information provided by the SaMD (i.e. to treat or diagnose; to drive clinical management or to inform clinical management) is defined by the significance of information provided by the SaMD to the healthcare decision. The state of healthcare situation or condition identifies the seriousness of the disease / condition ( i.e. critical; serious; or non-serious).
On the base of intended use and healthcare situation four classes of AI / ML-based SaMD are identified, as shown in the following table
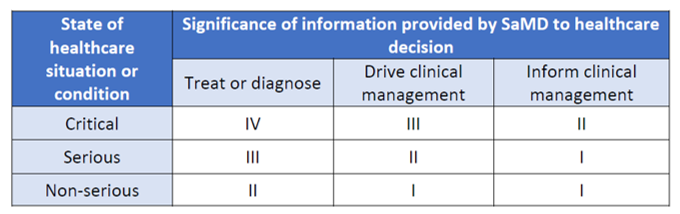
Table 1 – FDA Risk categorization of SaMD
[1] Reference document: Global Manufacturing Leadership Program - GMLP