
Validation of medical devices is concerned with the end product and – according to a FDA’s definition - it intends “to check that the device / system and its specifications conform with the user needs and the intended use(s)”
In its “Guidance on design validation” FDA provides recommendations for correct planning and running of the validation process of medical devices.
Here below the most relevant ones:
- Validation tests shall include production units under actual or simulated use conditions; In considering the intended use, it is relevant “to consider the intended users of the device and their qualifications and knowledge. Thus, the approaches to be applied in the case of devices intended to be used by healthcare professionals and laypersons should be different”;
- Design validation shall include risk analysis;
- The design validation process and its results shall be documented in the Design History File; it will include identification of the design, method(s), the date and the persons performing the validation;
- The device validation process should include the aspects related to
- packaging and labeling, as these elements are also vitally important for ensuring the use of a medical device in a safe and efficient manner;
- storing environment and transportation, including the impact of factors such as temperature, humidity, shock and vibration, corrosive atmospheres, etc.
- Depending on the complexity of the device and the risks associated with its use, clinical trials can be needed as part of the validation process; in such cases the medical devices have to be tested in the actual or simulated use environment;
- Apart from testing itself, additional methods of validation could be applied as well, e.g., a compilation of relevant scientific literature or provision of historical evidence.
Risk Management
FDA and other regulatory bodies give particular emphasis to risk assessment. A Risk Management Plan has to be foreseen; in it possible risks have to addressed and mitigation actions indicated for each of them. A Risk Management file must be prepared.
Addressed risks will be related to:
- Safety;
- failures and/or conditions impacting the effectiveness of the product for the intended use;
- data privacy and security; cybersecurity;
- risks related to the manufacturing process.
Three basic questions have to be answered:
- What can go wrong?",
- "What are the chances it could go wrong?",
- If it goes wrong what are the consequences?".
pFMEA (process Failure Mode & Effect Analysis) and the dFMEA (design Failure Mode & Effect Analysis) techniques could be used.
Validation at system level
The objective is to confirm that the product / system and its specifications conform to the intended use and fit the user's needs. The following diagram gives an overview of the tests to be conducted in the validation process.
Validation for software products
For software products or for products with functionalities highly depending on SW components additional tests are requested as indicated in the following diagram.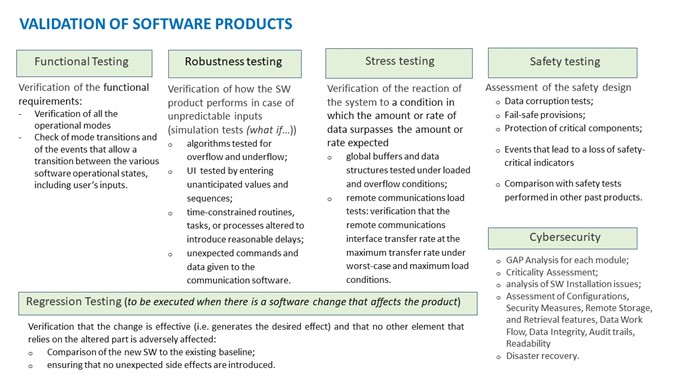
References